Methodenentwicklung
Bioinformatik
Workflows für die Biomarkerkandidaten-Detektion
Der biostatistische Workflow beginnt für die medizinischen Proben bereits mit der Planung der proteomischen Studien. Bevor Proben entnommen werden, wird die nötige Größe der gesamten Studie mit den Wissenschaftlern aller Kompetenzbereiche abgestimmt, um später statistisch signifikante Ergebnisse zu erlangen.
Sobald Proben vermessen wurden, beginnt der Auswerteworkflow für die Daten: zunächst werden die Proben mittels Qualitätskontroll-Metriken auf ihre Güte untersucht um mögliche Ausreißer möglichst frühzeitig zu erkennen. Das Aufzeichnen der Metriken ist ein automatisierter Prozess, bei dem sowohl Werte auf der rohen Messdatenebene zusammengetragen werden, als auch erste Identifikations- und Quantifizierungszahlen der Proben.
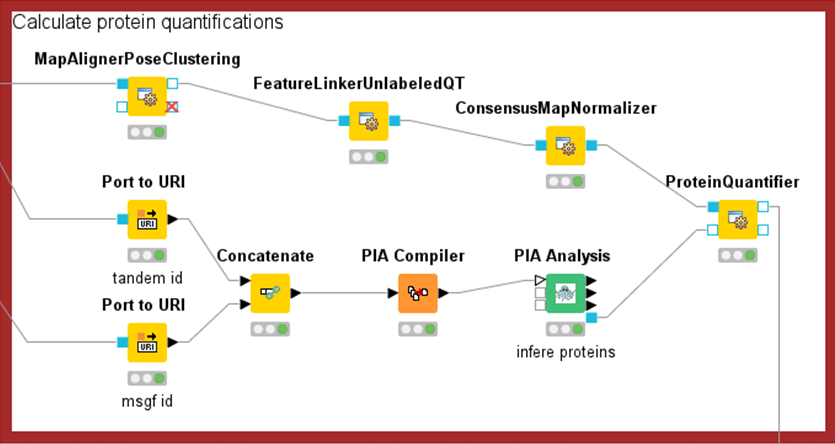
Die Messdaten werden dann mittels bioinformatischer Analysen ausgewertet. Hierbei müssen die Proteine einer Probe zunächst identifiziert werden. Bei Massenspektrometrie-basierten Methoden ist hierbei die korrekte Wahl von Protein-Sequenzdatenbanken und Schwellwerten für die Beschränkung von falsch-positiven Ergebnissen relevant. Neben der Identifizierung wird auch eine Proteinquantifizierung durchgeführt. Wie bei allen Hochdurchsatz-Messungen kann es bei der Proteomik zu geringfügigen systematischen Messungenauigkeiten kommen, die mit Normalisierungsmethoden ausgeglichen werden.
Sind die Daten entsprechend vorbereitet, können Methoden zur Biomarker-detektion angewendet werden. Hierbei werden univariate statistische Tests, aber auch Clustering-Methoden und Methoden des Maschinellen Lernens angewandt. Zuletzt werden die Biomarkerkandidaten in silico mittels Diskriminanz- und ROC-Analysen verifiziert. Hierdurch gefundene Kandidaten müssen dann noch durch Wetlab-Laborversuche validiert werden.
Optimierung von Methoden und vorhandener Auswerte-Software
Auf allen im Workflow genannten Ebenen werden bioinformatische Methoden kontinuierlich weiterentwickelt. Häufig ist auch die Anpassung einer etablierten Methode an die jeweilige Studie nötig, um qualitativ hochwertige Ergebnisse zu erhalten. Hierbei stellen auch immer neue technische Möglichkeiten der Massenspektrometer neue Herausforderungen. So sind die Datenmengen pro Messung in der Vergangenheit immer weiter angestiegen. Daraus resultierte eine effiziente Anpassung der bioinformatischen Methoden und Infrastruktur um diese Daten weiterhin effizient nutzen zu können. Aber auch durch die steigenden Genauigkeiten der Messverfahren sind ältere Methoden, z. B. zur Beschränkung der falsch-positiv-Rate von Peptid-Identifizierungen teilweise nicht mehr anwendbar, dafür können jedoch ganz neue Fragestellungen bioinformatisch analysiert werden.
Abbildung 2: Optimierung von Methoden zur Beschränkung der falsch-positiv-Rate

Deep Learning für die Detektion von Proteinvarianten
In den gewöhnlich zur Detektion von Proteinen und Peptiden verwendeten Proteinsequenz-Datenbanken werden nur die kanonischen Formen der Proteine gelistet. Zwar sind durch z.B. Genomsequenzierungsprojekte bereits viele Varianten und Mutationen von Proteinen bekannt, diese werden jedoch bisher nur selten für die Identifikation verwendet. Viele dieser bereits annotierten Varianten sind jedoch spezifisch für Krebs- und Neurodegenerationserkrankungen und deshalb für die Forschung im PRODI von besonderem Interesse.
Die Bioinformatik verfolgt Strategien, um diese Varianten in massenspektrometrischen Messungen detektieren zu können. Zum einen werden Verfahren entwickelt, um die große Anzahl an Varianten krankheitsspezifisch aus den Datenbanken zu extrahieren. Hierbei muss auch der bereits erwähnte verbesserte Ansatz zur Beschränkung der falsch-positiven Identifikationen berücksichtigt werden, da dieser immer dann besonders wichtig ist, wenn Suchräume besonders groß werden.
Zum anderen werden Methoden des Deep Learning angewendet, um eine sensitivere Datenbanksuche zur Spektrenidentifizierung zu entwickeln, sowie eine Methode zur de novo-Identifikation von Spektren. Für letztere sind keine Proteinsequenz-Datenbanken nötig, sondern mögliche Varianten und Mutationen werden erst in einem nachfolgenden Vergleich zu den Datenbanksequenzen ermittelt. Beide genannten Deep Learning Methoden helfen nicht nur dabei, bisher nicht detektierbare krankheitsspezifische Varianten und Mutationen nachzuweisen, sondern auch für Massenspektren die zugrundeliegenden Peptidsequenzen aufzuspüren, die bisher nicht identifizierbar waren und deshalb zur „dark matter“ der Proteomik gezählt werden.